

David R. Seaman, D.C. and James F. Winterstein, D.C.
Abstract:
Background and Objectives: Since the founding of the chiropractic profession, very few efforts have been made to thoroughly explain the mechanism(s) by which joint complex dysfunction generates symptoms. Save for a few papers, only vague and physiologically inconsistent descriptions have been offered. The purpose of this article is to propose a precise and physiologically sound mechanism by which symptoms may be generated by joint complex dysfunction.
Data Sources: The data was accumulated over a period of years by reviewing contemporary articles and books, and subsequently retrieving relevant papers. Articles were also selected from volumes 1-4 of the Chiropractic Research Archives Collection. The Nexus, published by the David D. Palmer Health Sciences Library, and In Touch, published by Logan College of Chiropractic Library, were reviewed and relevant articles were retrieved. Medline searches were found to be ineffective because appropriate key indexing terms were difficult to identify.
Data Synthesis: The symptoms generated by joint complex dysfunction, such as pain, nausea and vertigo, are probably caused by increased nociceptive input and/or reduced mechanoreceptive input.
Conclusions: Joint complex dysfunction should be included in the differential diagnosis of pain and visceral symptoms because joint complex dysfunction can often generate symptoms which are similar to those produced by true visceral disease.
Key Indexing Terms: Allodynia; Central Sensitization; Dysafferentation; Joint Complex Dysfunction; Mechanoreception; Nociception; Nociceptor Sensitization
INTRODUCTION
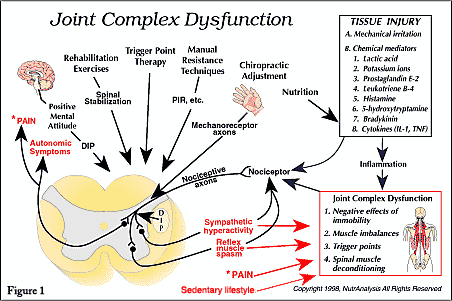
In a recent article, the term joint complex dysfunction was suggested as a replacement for subluxation complex [1]. Joint complex dysfunction refers to pathological and functional changes that occur in joint complex structures including (a) the negative effects of hypomobility/immobility, (b) functional imbalances such as muscle tightening or shortening and (c) myofascial trigger points. In short, the article demonstrated how the term joint complex dysfunction allows for a more descriptive and pathophysiologically precise discussion of spinal dysfunction compared with the term subluxation/subluxation complex. The author also proposed that the chiropractic profession adopt the term “dysafferentation” to describe the neuropathophysiological effects of joint complex dysfunction that act to generate symptoms [1].
The topic of symptom generation remains a source for debate within the chiropractic profession. For example, B. J. Palmer, who was often referred to as the “developer” of chiropractic, maintained “a peculiar belief in the perfection of the incoming (afferent) sensory system” [2]. In his writings, Palmer indicated that subluxations only affected efferent pathways and not the afferent system [2]. Many modern-day chiropractors still promote this notion. They believe that subluxations impinge upon spinal nerves at the level of the intervertebral foramina and interfere with the conduction of impulses innately generated within the brain and which subsequently pass through neural tissue, with the result that tissue supplied by the affected nerves could suffer some form of functional insult [3].
Another popular notion is that upper cervical subluxations or misalignments can somehow impinge upon the medulla and interfere with the transmission of “the mental impulse life force” [4]. To our knowledge, at the present time, there is no evidence to support such opinions about this type of relationship between joint complex dysfunction and efferent nerve function. However, as will be discussed in this article, a great deal of information suggests that joint complex dysfunction affects the afferent system by altering the function of nociceptors and mechanoreceptors found within the structures of the joint complex. The purpose of this article is to describe the sensory receptors and their relationship to afferent input and to describe possible symptoms that can develop in response to enhanced nociceptor input and reduced mechanoreceptor input, which has been referred to previously as dysafferentation [1]. We should mention that much confusion exists regarding sensory receptor terminology [5]. Consequently, a discussion about receptors and their function can lead to unnecessary arguments [6, 7]. For this reason and for the sake of clarity in general, pertinent terminology will be discussed in appropriate sections of this article.
DISCUSSION
Afferentation and Deafferentation
Afferentation refers to the transmission of afferent nerve impulses; deafferentation is defined as the elimination or interruption of afferent nerve impulses, as by destruction of the afferent pathway [8]. In neurological literature, the word “deafferentation” is typically reserved for conditions in which peripheral nerves are either damaged, completely severed or avulsed [9-11]. Because joint complex dysfunction is very rarely associated with peripheral nerve injury, it is not appropriate to use the word deafferentation.
Dysafferentation
Dysafferentation refers to an imbalance in afferent input such that there is an increase in nociceptor input and a reduction in mechanoreceptor input [1]. Notice that proprioceptors are not mentioned; this is because contemporary texts do not consider proprioceptors a category of receptor. A recent article explained why ‘proprioceptor’ is actually an obsolete, inaccurate word [5].
According to standard texts in neurology, there are two categories of somatic receptors: nociceptors and mechanoreceptors [5, 12-14]. At the present time, an emerging body of research indicates that abnormal joint complex function can alter the activity of nociceptors [15-19] and mechanoreceptors [20-23], such that nociceptive activity increases and mechanoreceptive activity decreases. Many authors and researchers involved in joint adjusting and manipulation realize this and use the terms “altered afferent input,” “abnormal afferent input” or similar terms when discussing the neuropathophysiological component of joint complex dysfunction [24-32]. For example, Peterson stated that, “somatic dysfunction and/or joint dysfunction induce persistent nociceptive input and altered proprioceptive input” [26]. Peterson and Bergmann described vertebral joint dysfunctions and their associated mechanical alterations, pain and potential local inflammation as lesions capable of inducing chronically altered nociceptive and proprioceptive input” [27]. Hooshmand illustrated how restricted joint mobility results in decreased firing of large diameter mechanoreceptor axons (A-beta fibers) and increased firing of nociceptive axons (A-delta and C fibers) [20]. Henderson used the term “altered somatic afferent input theory” to classify a neurophysiologic theory of chiropractic subluxation [28]. He proposed that chiropractic adjustment might normalize articular afferent input to the nervous system, which reestablishes normal nociceptive and kinesthetic reflex thresholds.
The information in the previous paragraph demonstrates that researchers in different professions have acknowledged the fact that compromised joint function will alter afferent input, such that nociception is enhanced and mechanoreception is reduced. In the biomedical literature, the prefix “dys” is used to describe activity that is abnormal, bad, difficult or disordered [33]. For this reason, we propose that the chiropractic profession adopt the word ‘dysafferentation’ to describe the abnormal afferent input associated with joint complex dysfunction [1].
The remainder of this article will discuss potential symptoms that may develop as a consequence of dysafferent input (i.e., increased nociception and reduced mechanoreception). To accomplish this task, both the neuroanatomy and physiology related to nociceptors and mechanoreceptors will be discussed and then related to symptom generation.
Nociceptors
Neuroanatomy
Many believe that spinal tissue nociceptors can be found in:
- skin,
- subcutaneous and adipose tissue,
- fibrous capsules of the apophyseal and sacroiliac joints,
- spinal ligaments,
- the periosteum covering vertebral bodies and arches (and attached fascia, tendons and aponeurosis),
- the dura mater and epidural fibro-adipose tissue,
- the walls of blood vessels supplying the spinal joints, sacroiliac joints and the vertebral cancellous bone,
- the walls of epidural and paravertebral veins and
- the walls of intramuscular arteries and at least the outer third of the annulus fibrosis [34-41].
Wyke provides the most vivid anatomical description of the nociceptive receptor system [41]. He described interstitial nociceptors as “a continuous tri-dimensional plexus of unmyelinated nerve fibers that weaves (like chicken-wire) in all directions throughout the tissue.” A similar plexus of unmyelinated nerve fibers is embedded in the adventitial sheath and encircles each blood vessel. Commenting on nociceptive C fiber innervation, Charman stated that the “network of each C fibre innervates a three-dimensional receptive field of between 6 and 15 mm in diameter and of variable depth with extensive field overlapping between adjacent C-fibers” [37]. Thus, we can envision the presence of an almost unending meshwork of nociceptors within the various tissues described earlier.
Nociceptors are classified as mechanical nociceptors, mechanothermal nociceptors and polymodal nociceptors, depending on the type of energy used to activate them in the nociceptive range. Polymodal nociceptors are activated by noxious mechanical and thermal stimulation, as well as by chemical mediators released from the injured tissues [42].
Recent research has demonstrated the presence of articular nociceptors with thresholds so high that they cannot be excited by acute noxious stimulation, for example, mechanical injury to the joint [17-19, 43]. Thus, these nociceptors are normally mechano-insensitive and have been characterized as solely chemosensitive [43]. This special category of nociceptors is called silent or sleeping nociceptors. Sleeping nociceptors are thought to awaken in the presence of chemical mediators released from injured tissues [17, 18, 43], at which time they become mechanosensitive. It is thought that the activation of these afferents may not only represent an extra source of nociceptive input but may also be important in promoting central sensitization [43].
Generally speaking, mechanical and mechanothermal nociceptors are innervated by A-delta axons, whereas polymodal and silent nociceptors receive their innervation from C fibers. Within the spinal cord, nociceptive afferents send collaterals to supra- and infra-adjacent segments. For example, A-delta fibers spread collaterals three to six segments rostrally and an equal number caudally, whereas C fibers spread two to three (or possibly more) segments above and below the level of entry [44].
A recent review by Charman provides a vivid description of the central connections of nociceptive afferents [37]. Nociceptive afferents travel up the anterolateral system and ultimately terminate in the spinal cord, a variety of brainstem nuclei, the limbic system, frontal lobes, parietal lobes, insula cortex and temporal lobes.
Peripheral nociceptive sensitization
Nociceptors have high thresholds of activation, which means that, under normal circumstances, only stimuli that are either potentially or overtly tissue-damaging can depolarize a nociceptor [45, 46]. Normally, light touch and normal joint motion cannot depolarize a nociceptor [5].
Although nociceptors normally have high thresholds, certain physiological environments can result in the lowering of nociceptive thresholds, such that light touch and normal movement patterns can cause nociceptor depolarization and excitation of nociceptive afferent pathways. Peripheral sensitization is the term used to describe the process by which nociceptor thresholds are lowered [47], thus enhancing the transmission of nociceptive impulses into the spinal cord. The driving force behind the sensitization process seems to be the chemical mediators released after tissue injury. Prostaglandin E-2, leukotriene B-4, bradykinin, histamine and 5-hydroxytryptamine are thought to be the main chemical mediators that can sensitize nociceptors [10, 47]. Local tissue acidity is also thought to be capable of participating in the sensitization process [47 49].
Recent research suggests that norepinephrine release from sympathetic terminals into the area of tissue injury can also sensitize nociceptors [47, 50-52]. The exact mechanism by which this occurs is not well understood. It is possible that norepinephrine released from sympathetic terminals activates alpha-1-adrenergic receptors found on the nociceptor membrane [50]. It is also possible that sympathetic discharge into the area of tissue injury promotes the release of prostaglandins [53].
It is not clear whether substance P is directly involved in the sensitization process [10, 47, 54]. We know that substance P is produced in the dorsal root ganglion cells and then transmitted to the spinal cord and to the nociceptor. Substance P is released from nociceptors after they are activated by noxious input. The activity of substance P in this local environment can cause further accumulation of bradykinin and the release of histamine from mast cells and 5-hydroxytryptamine from platelets, all of which can promote the sensitization of local nociceptors [10].
It is thought that chemical mediators depolarize and/or sensitize a nociceptor through their interaction with chemosensitive receptors on the membrane of the nociceptor, which influences the flow of sodium, calcium and potassium ions Some of the chemical mediators have receptors linked directly to ion channels, whereas the receptors for other mediators are linked to second messenger systems that modulate ion channels. For more details, see Rang et al. [53].
It is important to consider the degree to which nociceptor sensitization can influence the activity of the associated afferent fibers and the spinal cord. Hanesch et al. provide a vivid example of afferent fiber function after nociceptor sensitization [17]. They studied the medial articular nerve in cats and discovered that, in normal joints, the afferent volley during a simple flexion movement comprises approximately 4,400 impulses per 30 seconds, including resting discharges. During inflammation, which promotes nociceptor sensitization, the afferent volley comprises some 30,900 impulses per 30 seconds, which represents about a sevenfold increase compared with normal conditions. They indicated that, “in individual afferents that have been studied consecutively under both normal and inflamed conditions, the afferent discharges sometimes increased more than 100-fold.”
It is thought that an increased nociceptive barrage caused by sensitized nociceptors plays a role in the development of central nociceptive sensitization. Researchers have yet to discover all of the details about central sensitization. Nonetheless, we will attempt to provide the most relevant data in the next section.
Central nociceptive sensitization
After tissue injury, nociceptors exhibit spontaneous activity, lowered thresholds and increased responsiveness to noxious stimuli, which leads to hyperexcitability and altered neuronal processing in the spinal cord and brain [55]. The term ‘central sensitization’ refers to this increased excitability of nociceptive neurons in the central nervous system (CNS) [56]. Of importance to the chiropractic profession is the fact that joint nociceptors, as with nociceptors in other tissues, can be sensitized [57]. In addition, joint and muscle nociceptors are much more capable of producing central nociceptive sensitization than cutaneous nociceptors [58].
In 1942, Denslow and Hassett reviewed the literature and demonstrated that the concept of spinal cord hyperexcitability had been around since at least the 1930s [30]. At that time, the term central excitatory state was used to describe central sensitization. Denslow and Hassett were the first to suggest and demonstrate that spinal dysfunction (i.e., joint complex dysfunction) was associated with central sensitization. They credited Charles Sherrington for developing the concept of the central excitatory state. They state that the evocation of additional activity by a stimulus which, in the normal would be ineffective, is explained by Sherrington’s concept of a motoneuron pool in which there is an enduring subliminal central excitatory state (CES) created by sub-threshold stimuli [30].
In 1947, Denslow et al. used the terms facilitation and central facilitation in an effort to describe what is now referred to as central sensitization [59]. Recently, Patterson indicated that Irvin Korr is credited with coining the term facilitated segment [60], about which Korr wrote numerous articles [61 -67].
In 1983, and apparently without knowledge of the work by Denslow et al., Woolf hypothesized and then experimentally demonstrated the presence of a hyperexcitable central component in post-injury pain hypersensitivity [68]. In 1987, Woolf provided a clear explanation of central sensitization: “C fiber input to the spinal cord, in addition to producing input concerning the onset, location and duration of the peripheral noxious stimuli, also produces a prolonged increase in the excitability of the spinal cord” [58]. Research demonstrated that brief conditioning stimuli of nociceptive C -fibers (up to 20 sec) at low frequencies (1 Hz) can produce a prolonged excitation (up to 90 minutes) of the spinal cord. Additionally, it was shown that C fibers innervating deep structures, such as joints or muscles, can more readily produce central facilitation than can cutaneous C fibers – [58]. The activation of nociceptive C afferents can also produce profound changes in the receptive field properties of dorsal horn neurons, such as an expansion of the receptive field size, an increase in spontaneous activity, and an increase in response to innocuous stimuli [58].
Since Woolf’s initial article in 1983, a great deal of research has been devoted to understanding the process of central sensitization [55, 56, 69-81]. Most authors now agree that central sensitization manifests in CNS neurons as increased spontaneous activity, reduced thresholds or increased responsiveness to afferent inputs, which are prolonged after discharge from repeated stimulation, and expanded receptive fields of -dorsal horn neurons [56]. Many think that these CNS changes are caused by an increase in excitatory inputs and/or a loss of inhibitory inputs, which result in a net excitation of the dorsal horn [69].
At the present time, an emerging impression is that spinal cord plasticity is responsible for the development of central sensitization [55, 56, 69-71, 82]. In general, plasticity refers to an adaptation of the nervous system in response to changes in the associated internal or external environment [83]. Kandel states-that plasticity is a “change in the effectiveness of specific synaptic connections” [84]. Research suggests that the physiological basis of plasticity involves an increase in gene expression, particularly intermediate-early genes, such as c-fos and c-jun [70, 85]. With respect to central sensitization, plastic changes begin after the release of various excitatory transmitter substances from nociceptive afferents, such as substance P, calcitonin gene-related peptide, aspartate and glutamate, and their subsequent action at N-methyl-D-aspartate receptor sites. Dubner and Ruda provide a review of this proposed mechanism [70].
It is thought that central sensitization extends to neurons in the dorsal and anterior horns of the spinal cord, the thalamus and even higher centers [56, 86, 87]. In other words, noxious input leads to the hyperexcitability of alpha-motoneurons, preganglionic sympathetic neurons, spinal cord nociceptive projection neurons, thalamic projection neurons and other neurons in the brain. A variety of symptoms and conditions can develop in response to these changes. For example, it is known that nociceptor sensitization and central nociceptive sensitization of projection neurons causes increased pain [55]. From a clinical perspective, chiropractors routinely encounter pain associated with a sensitized nociceptive system. It is common to discover that gentle or normal palpation of spinal tissues results in the experience of pain. The word allodynia is used to describe this state, in which normally painless stimuli result in pain [47].
Considering the fact that nociceptive input reaches subcortical areas, such as the brainstem hypothalamus [37], it is also likely that -a wide variety of neuro-endocrine responses and seemingly unrelated symptoms could develop in response to a sensitized nociceptive system. The following two sections discuss these relationships in some detail.
Neuro-endocrine responses caused by nociceptive input to subcortical centers
We know that nociceptive afferent input travels up the anterolateral pathway, which contains the spinoreticular and spinothalamic tracts. -Bonica provides a succinct explanation of the neuroendocrine responses associated with such activity [87]:
Suprasegmental reflex responses result from nociceptive induced stimulation of medullary centers of ventilation and circulation, -hypothalamic (predominantly sympathetic) centers of neuroendocrine function and some limbic structures. These responses consist of hyperventilation, increased hypothalamic neural sympathetic tone and increased secretion of catecholamines and other catabolic hormones. The increased neural sympathetic tone and catecholamine secretion add to the effects of spinal reflexes and further increase cardiac output, peripheral resistance, blood pressure, cardiac workload and myocardial oxygen consumption. In addition to catecholamine release, there is an increased secretion of cortisol, adrenocorticotrophic hormone, glucagon, cyclic AMP, antidiuretic hormone, growth hormone, renin and other catabolically acting hormones, with a concomitant decrease in the anabolically acting hormones insulin and testosterone.— Cortical responses, in addition to and including the perception of pain as an unpleasant sensation and negative emotion, initiate the psychodynamic mechanisms of anxiety, apprehension and fear. These, in turn, produce cortically mediated increases in blood viscosity, clotting time, fibrinolysis and platelet aggregation. Indeed, cortisol and catecholamine responses to anxiety usually exceed the hypothalamic response that is provoked directly by nociceptive impulses reaching the hypothalamus.
Bonica demonstrates the degree to which nociceptive input can metabolically compromise the host. As implied above, subcortical responses can occur with or without the experience of pain. Perhaps such asymptomatic neuroendocrine responses, induced by nociceptive input, play a role in the pathogenesis of degenerative diseases, such as cardiovascular disease, cancer, diabetes, -arthritis and Alzheimer’ 5 disease. We need more research in this area, which becomes more obvious when the devastating effects of hypercortisolemia are considered.
Cortisol levels are increased by nociception, pain, inflammation, trauma, anxiety, fear, apprehension, prolonged and strenuous exercise and hypoglycemia [87, 88]. When stressors are present for protracted periods of time, feedback suppressibility of cortisol can be impaired [88].
The damaging effects of excess endogenous cortisol are as jeopardizing as those associated with exogenous intake in the form of medication. Such a relationship can be better appreciated when we understand that -cortisol secretion can rise 20-fold when the adrenal gland is chronically stimulated [88].
Excess cortisol produces a continuous drain on body protein stores, most notably in muscle, bone, connective tissue and skin. Hypercortisolemia causes a variety of tissue-specific changes, including a reduction in rapid-eye-movement sleep; a reduction of cell-mediated immunity by inhibiting the production of interleukin- 1, interleukin-2 and gamma-interferon; a decrease in the proliferation of osteoblasts; and a reduction in collagen synthesis. We know cortisol antagonizes the action of insulin, which results in decreased glucose use. Cortisol stimulates lipogenesis in specific body locations, including the abdomen, trunk and face [88].
Chronic hypercortisolemia may play a role in spinal muscle degeneration. We know that cortisol preferentially reduces the ratio of slow oxidative type I muscle fibers to fast glycolytic type Il-B -fibers [88]. This may enhance the deconditioning -of spinal muscles that occurs as a consequence of sedentary living and aging and may promote spinal injury and chronic joint complex dysfunction.
There are also a number of diseases that are driven by hypercortisolemia. Dilman and Dean actually characterize hypercortisolemia as a disease [89]. They coined the term hyperadaptosis to describe a state characterized in its latent stage -by an excessive and -prolonged elevation of cortisol levels in response to stressors, and in its overt stage by an elevation of basal cortisol levels in the absence of apparent stressors. Several conditions are known to develop as a consequence of hypercortisolemia, including heart disease, various cancers, hypertension, depression, obesity and diabetes [89].
Symptoms caused by nociceptive input to subcortical centers
Feinstein et al. were the first to clearly describe some symptoms associated with noxious irritation of spinal tissues [90]. They injected hypertonic saline into interspinous tissues and paraspinal muscles of normal volunteers for the purpose of characterizing local and referred pain patterns that might -develop. What they discovered was surprising:
The pain elicited from muscles was accompanied by a characteristic group of phenomena which indicated involvement of other than segmental somatic mechanisms. The manifestations were pallor, sweating bradycardia, fall in blood pressure, subjective ‘faintness,” and nausea, but vomiting was not observed. Syncope occurred in two early procedures in the series of paravertebral injections and was subsequently avoided by quickly depressing the subject’s head or by having him lie down at the first sign of faintness. These features were not proportional to the severity of or to the extent of radiation; on the contrary, they seemed to doimnate the experience of subjects who complained of little pain, but who were overwhelmed by this distressing complex of symptoms.
Feinstein et al. -referred to these symptoms as autonomic concomitants [90]. It is likely that these autonomic concomitants were caused by nociceptive stimulation of autonomic centers in the brainstem, particularly the medulla [87]. Fein stein et al. indicate that “this is an example of the ability of deep noxious stimulation to activate generalized autonomic responses independently of the relay of pain to conscious levels” [90]. In other words, pain may not be the symptomatic outcome of nociceptive stimulation of spinal structures. Such a conclusion has profound implications for the chiropractic profession. Clearly, patients do not need to be in pain to be candidates for spinal adjustments.
Nansel and Szlazak published the most recent article devoted to autonomic concomitants associated with nociceptive input – [91]. They explained -that it is now well-established that nociceptive input from somatic and visceral structures “converges on common pools of interneurons within the spinal cord and brainstem.” As -a consequence, nociceptive input from somatic structures can “create complex patterns of signs and symptoms -that can often be virtually identical to and, therefore, easily mistaken for those induced by primary visceral disease.” They have collected more than 200 scientific articles that deal with various somatic visceral disease mimicry syndromes.
In summary, there are many neuroendocrine and -symptomatic presentations that can occur in response to nociceptive input. It is very likely that no two patients will present in the exact same fashion, even if joint complex dysfunction exists in the same spinal location. The symptomatic picture can become even more complex when the consequences of reduce mechanoreception -are considered.
Mechanoreceptors
Mechanoreceptors are located in the skin, muscles, joint structures and the intervertebral disc [13, 14, 92]. Examples of mechanoreceptors include muscle spindles, golgi tendon organs (GTOs) and a variety of corpuscular mechanoreceptors, such as Ruffini endings, Merkel cell complexes, Meissner’s corpuscles, Pacinian corpuscles and many others [14].
Many believe that, as a group, mechanoreceptors respond only to weak mechanical stimuli, such as touch and joint movement, and not to nociceptive stimuli with higher frequencies [l8, 19, 42, 93]. -As would be expected, several authors suggest that reduced joint movement results in less mechanoreceptor activation [20-23]; however, the degree to which mechanoreceptor input would be compromised by joint hypomobility is unknown at the present time. Although it may be difficult to quantify such mechanoreceptor deficits in the laboratory, research suggests that a reduction in mechanoreceptor afferent input can result in the development of symptoms that can be identified in the clinical setting. For example, de Jong et al. injected human subjects with lidocaine in the area halfway between the mastoid process and carotid tuberde at the level of the second and third cervical vertebrae [94]. Injections were made unilaterally. Immediately after injection, symptoms of dysequilibrium began to appear. Symptoms included ataxia, hypotonia of the ipsilateral arm and leg and a strong sensation if ipsilateral falling or tilting. The symptoms were more pronounced on the side of injection and lasted for about an hour. The authors suggested that the injection of local anesthetics interrupts the flow of afferent information from neck and muscle receptors,” which can affect vestibular nuclei function and promote a variety of vestibular symptoms [94]. Presumably, the receptor types to which de Jong et al. refer include corpuscular mechanoreceptors, muscle spindles and GTOs.
A brief review of mechanoreceptor subtypes, their basic functions and the neuroanatomical pathways associated with mechanoreceptors will help to outline potential symptoms that may develop because of joint complex dysfunction.
Corpuscular mechanoreceptors
Corpuscular mechanoreceptors are found in the skin, joint structures and muscles [14, 21, 95]. in general, -people believe that corpuscular mechanoreceptors are associated with A-beta fibers [12, 13].
Mechanoreceptor afferents (A-beta fibers) influence the nervous system in many ways. For example, at the spinal cord level, mechanoreceptor input can inhibit nociception [14, 20, 96-99]. Thus, it is very likely that reduced mechanoreceptive activity will enhance the nociceptive input associated with joint complex dysfunction. Also, mechanoreceptor afferents can reduce sympathetic hyperactivity [20, 99, 100]. Thus, it is reasonable to suggest that reduced mechanoreception will enhance segmental sympathetic hyperactivity and somatomotor output.
Clearly, mechanoreceptor afferents have important functions in the CNS. It seems that it would be ideal to have an abundance of mechanoreceptors functioning at an optimal level at all times. Unfortunately, it also seems that there are relatively few corpuscular mechanoreceptors compared with nociceptors.
The precise concentration of corpuscular mechanoreceptors in somatic tissues is unknown. Recent research suggests that the concentration of nociceptors far exceeds that of corpuscular mechanoreceptors. Schmidt et al. discus-s the percentage of fiber types in the medial articular nerve and posterior articular nerve of the cat [19]. Only 9% of the fibers in the medial articular nerve and 26% in the posterior articular nerve were mechanoreceptive. Such a low percentage of mechanoreceptive afferents suggests that maintaining prQper joint mobility is very important.
Muscle spindles
Muscle spindles are classically described as receptors that send information into the CNS about muscle length or the rate of change of muscle length. It is generally believed that muscle spindles lie parallel to extrafusal fibers in the center of a muscle. Muscle spindles have primary endings associated with a group Ia afferent fiber and secondary endings associated with a group II afferent fiber. Both group Ia and II fibers are thought to have numerous connections within the spinal cord.
Group Ia afferents are involved in several cord reflexes that modulate extrafusal muscle function, such as the stretch reflex, recurrent inhibition, reciprocal inhibition and the cross extensor reflex [101]. All of these reflexes are very important for promoting smooth and controlled movements. Group II afferents are not as well described as Ia afferents. It is generally believed that group II afferents primarily affect the static component of the stretch reflex.
In general, the greatest concentration of muscle spindles are located in muscles involved in fine movements and posture, whereas the lowest concentration is found in muscles involved in gross movement [102]. Research has demonstrated that the digits and neck contain the greatest density of muscle spindles. Indeed, it has been stated that the neck muscles contain a “bewildering number of muscle spindles” [103]. Dvorak and Dvorak indicate that, per gram of muscle tissue, the rectus femoris contains 50 muscle spindles, whereas the suboccipital muscles contain approximately 150-200 muscle spindles and the intertransverse muscles in the cervical spine contain between 200-500 muscle spindles [99]. Some have suggested that the intertransverse muscles of the neck and lower back may actually function as mechanoreceptors and not as muscles [104].
The distribution of muscle spindles within muscles is far more varied and complex than the classic description. Richmond et al. indicated that muscle spindles can be associated with one another in several ways, including:
(a) paired associations, in which two or more spindles lie side-by-side,
(b) parallel associations, in which two or more intrafusal fiber bundles are contained within a common capsule for some part of their -length and
(c) tandem associations, in which two or more spindle units are linked in series by a common intrafusal fiber that runs through each spindle unit in succession.”
It is not uncommon to find muscle spindles linked in complex arrays that span the length of an intervertebral muscle. Some spindles are also found in close contact with Paciniform corpuscles and GTOs [103].
GTOs
GTOs are usually described as muscle tension receptors. Many believe that GTOs lie in series with extrafusal fibers. In other words, GTOs are found at the junction of a muscle and its tendon.
GTOs are stimulated when muscle contraction generates tension in a muscle. The frequency of firings increase in proportion to the increasing muscle tension [103]. GTOs are innervated by a group lb afferent fiber. The lb afferent enters the cord and excites an interneuron located in the intermediate region of spinal cord gray matter. This so-called lb inhibitory interneuron serves to inhibit the alpiha-motoneuron that innervates the same muscle that was contracted and created the tension. It should be mentioned that the lb inhibitory intemeuron receives convergent input from -Ia afferents from muscle spindles and A-beta afferents from cutaneous and joint receptors [100, 105].
GTOs are highly concentrated in neck muscles. Their distribution tends to be nonuniform. They are found -along internal aponeurosis and where muscles attach to the vertebral process. In both the deep and more superficial neck muscles, spindles and -GTOs are often clustered in complex receptor arrays [103].
In recent years. it has been shown the GTOs have a dynamic sensitivity and are more suited to signaling rapidly changing tensions rather than static levels of tension. Research has shown that GTOs respond to forces as low as 4 mg.
Supraspinal connections
We know that afferent input from corpuscular mechanoreceptors, muscle spindles and GTOs can influence brain -function. Researchers have stated that mechanoreceptive input is partially responsible for proprioception [14, 96] and suprasegmental motor control [106]. Indeed, it seems that many supraspinal centers depend on afferent input, including the cerebellum and cerebral cortex. For example, Carpenter states that afferent fiber input to the cerebellum exceeds efferent fibers by a ratio of-approximately 40:1, which demonstrates the degree to which afferent input is needed by the CNS [107]. With this information in mind, it is important to consider -the probability that joint complex dysfunction is associated with the degeneration, atrophy and deconditioning of mechanoreceptor rich tissues, such as muscles and joint structures [1, 107-109].
The following sections describe the main afferent and subsequent efferent connections of the cerebellum and cerebral cortex. Based on these connections and related research findings, a variety of potential symptoms that may manifest in response to the reduced mechanoreceptive input associated with joint dysfunction will be described.
Cerebellum
Brodal provides a detailed description of mechanoreceptive input to the cerebellum [95]. Afferents enter the cerebellum through all three cerebellar peduncles. Whereas the superior peduncle contains mostly efferent fibers and some afferent fibers, the middle peduncle contains only afferent fibers and the inferior peduncle contains mostly afferent fibers and some efferent fibers. The discussion that follows focuses on afferent information that travels through the middle and inferior peduncles.
Afferents entering the middle cerebellar peduncle come from the cerebral cortex via the corticopontocerebellar tract, which contains some 20 million fibers [95]. The majority of these fibers project to the lateral cortex of the cerebellum, which is involved in the coordination and regulation of sequential and volitional motor activities initiated by the cerebral cortex [95, 110]. Clearly, without afferent input, the lateral cortex could not properly modulate motor control.
The lateral cortex is often called the cerebrocerebellum because it receives input exclusively from the cerebral cortex by way of the pontine nuclei [111]. The heaviest projections come from the primary motor area (Brodmann area 4), the primary sensory area (Brodmann areas 3, 1 and 2), a somatosensory association area (Brodmann area 5) and from parts of the visual areas related to the peripheral visual field [95]. Areas 3, 1 and 2, the primary sensory area, each receive afferents from specific receptors [107]. Group Ia muscle spindle afferents project to area 3a. Cutaneous afferents project to area 3b. Joint afferents project to area 2. Area 1 receives input from both cutaneous and deep tissue receptors. This information makes it clear that a significant level of mechanoreceptor afferent input indirectly reaches the cerebellum by way of the cerebral cortex.
The inferior peduncle contains some 0.5 million fibers [95], most of which originate from receptors in axial and appendicular structures. -Afferents from spinal structures end in the cerebellar vermis, whereas afferents from the extremities end in the intermediate cortex, which is also referred to as the paravermal region. It is important to note that this input is somatotopically organized. Nolte indicates that the principal sources of afferent input to the vermis and intermediate cortex come via the -spinal cord from mechanoreceptors in the skin, muscles and joints [110]. For this reason, the vermis and intermediate cortex are often referred to as the spinocerebellum. It should be mentioned that both the vermis and intermediate cortex also receive afferent input from the motor cortex by way of the corticopontocerebellar tract. This input is somatotopically organized so that the cortical fibers end in the same pattern as those from mechanoreceptor afferents [1-10].
As far back as 1960, researchers determined that the dorsal spinocerebellar tract conveys information to the vermis and intermediate cortex from muscle spindles, GTOs and corpuscular mechanoreceptors in the skin. Later, it was determined that the dorsal spinocerebellar tract also conveys information from joint receptors [95]. We know that the dorsal spinocerebellar tract conveys mechanoreceptive information from the lower half of the body and the cuneocerebellar tract relays mechanoreceptor afferent input from the upper half [95, 107]. The importance of the spinocerebellar pathways is demonstrated by the conduction velocity of its component fibers. We know that impulses are transmitted at velocities up to 120 rn/sec for the purpose of instantaneously apprising the cerebellum of peripheral movements [12]. Additional afferents that travel through the inferior cerebellar peduncle to end in the spinocerebellum arise from the inferior olive and central-cervical nucleus.
The inferior olive also provides input to the cerebellum from mechanoreceptor afferents that end somatotopically in the olive [95]. Carpenter [107] indicated that afferents from cutaneous receptors and lb afferents from GTOs contribute to the spino-olivary tract. Proske indicated that the principal supraspinal site of termination for OTO afferents is the cerebellum [105]. The major -projection is from the spinocerebellar tracts and the secondary projection comes from the inferior olive via the spino-olivary tract [105]. If the spino-olivary tract fis consistent with other ascending mechanoreceptive tracts, it is likely that all types of mechanoreceptor afferents project to the inferior olive. Brodal states that “the quantitatively most important contingents of afferents mediate spinal impulses” [95].
The inferior olive is a very important structure and far too complex to discuss in this article. In brief, we know that the inferior olive projects -to all parts of the cerebellar cortex and all deep cerebellar nuclei (i.e., the dentate, globus, emboliform (nucleus interpositus) and fastigial). There are even reciprocal connections between each cerebellar nucleus and the olive. In addition to mechanoreceptor afferents, the inferior olive also receives -input from the cerebral cortex (chiefly the motor cortex), red nucleus, mesencephalic reticular formation, superior colliculus and pretectum [95]. The symptoms associated with experimental oblation of the inferior olive are similar to those associated with destruction of the entire contralateral cerebellum [110].
The central cervical nucleus (CCN), located in lamina VII of the first four cervical segments, projects to the cerebellar vermis [95]. Originally, the CCN was thought to receive only upper cervical mechanoreceptor afferents [95, 112]. Recent research suggests that the CCN may receive mechanoreceptor afferents from as low as the lumbar spine [113]. However, there is evidence to suggest that the CCN is most powerfully influenced by receptors in deep neck muscles [114].
The vermis functions to control equilibrium, posture, muscle tone and locomotion [12, 95, 107]. Ghez states that the vermis is involved in axial and proximal motor control [111]. It must be remembered that, without proper afferent input, these functions would be compromised.
Assuming that the vermis receives sufficient mechanoreceptive input, it will modulate motor function by projecting to a variety of nuclei. The vermis projects somatotopically organized information to the fastigial nucleus, which then projects bilaterally to the lateral vestibular nucleu and reticular formation nuclei [111], particularly the nucleus reticularis pontis caudalis and the nucleus reticularis gigantocellularis [95]. The tracts associated with these nuclei include the lateral vestibulospinal tract, the medial reticulospinal tract and the lateral reticulospinal tract. respectively. We know the fastigial nucleus also projects to the thalamus and, ultimately, to the motor cortex [95, 111].
The intermediate cortex mainly coordinates the actions of the distal extremities [95, 107, 111]. It accomplishes this task by projecting to nucleus interpositus, which projects mainly to the red nucleus and motor cortex, affecting the rubrospinal tract and corticospinal tract, respectively. Many believe that the intermediate cortex compares commands emanating from the motor cortex with the actual position and velocity of the moving part (it receives this information from mechanoreceptors); then, by way of the nucleus interpositus, the intermediate cortex issues correcting signals [110]. Without adequate mechanoreceptor input, the ability of the intermediate cortex to modulate motor control would be compromised.
Thus far, both the cerebrocerebellum and spinocerebellum have been discussed. The third division of the cerebellum is known as the vestibulocerebellum, so named because it receives afferent input from the vestibular nerve and vestibular nuclei. The flocculonodular lobe is the specific area of the cerebellum referred to as the vestibulocerebellum. Afferents to the vestibulocerebellum also travel through the inferior cerebellar peduncle.
Most texts indicate that only vestibular afferents end in the flocculonodular lobe [11, 110, 111]; however, this is not the case. Guyton alluded to the fact that mechanoreceptor afferents from the neck also gain access to the flocculonodular lobe [12]. He explains that mechanoreceptors from the neck and body transmit information directly into the vestibular nuclei and indirectly, by way of the cerebellum, into the flocculonodular lobe. Apparently, mechanoreceptor input from the neck transmits signals that oppose the signals from the vestibular apparatus, which prevents a person from developing dysequilibrium when the head is laterally flexed or rotated. Guyton further stated that, “by far the most important proprioceptive information needed for the maintenance of equilibrium is that derived from the joint receptors of the neck” [12]. Although not described by Guyton, several other authors describe a specific relay nucleus by which mechanoreceptor afferents gain access to the vestibulocerebellum [95, 113, 114].
Brodal states that mechanoreceptor afferents project to the flocculonodular lobe via the group X nucleus located in the region of the vestibular nuclei [95]. Bakker and Abrahams explain that the neurons of group X seem to serve as a primary relay for cervical mechanoreceptor afferent input to the cerebellum [114]. Research also suggests that thoracic and lumbar mechanoreceptor afferents project to group X [113].
In summary, the flocculonodular lobe receives input from the vestibular apparatus in the inner ear and mechanoreceptor afferents, and then projects ipsilaterally to the middle, superior and inferior vestibular nuclei. Relatively few fibers project to the lateral vestibular nucleus [95]
Most authors agree that the medial vestibulospinal tract originates in the medial vestibular nucleus and descends bilaterally into the cervical and perhaps upper thoracic cord [95, 110, 111]. Ghez stated that, “this tract participates in the reflex control of neck movements so that the position of the head can be maintained accurately and is correlated with eye movements” [111].
The medial longitudinal fasciculus (MLF) is believed to originate in the superior and medial vestibular nuclei. Fibers in the MLF project to abducens. trochlear and oculomotor nuclei. which allow a person 5 gaze to stay fixed on an object while the head is moving [110]. This is known as the vestibulo-ocular reflex; Maeda has demonstrated that the cervical spine mechanoreceptors participate in this reflex by projecting to the group X nucleus [115].
The MLF also projects to the interstitial nucleus of Cajal [95]. It is thought that this nucleus probably plays an important role in mediating the effects of optic and vestibular impulses on the neck and body musculature and may also influence certain central effects on the autonomic system [95].
The vestibular nuclei also project to the cerebral cortex via both direct and indirect pathways. Animal experiments have revealed that within the facial region of the primary sensory area, there is a marked convergence of vestibular and somatosensory impulses arising from muscle spindles and from mechanoreceptors in the skin and joints [95]. These connections may exist in humans; Nolte indicates that each superior vestibular nucleus sends projections bilaterally to the facial region of the primary sensory area [110]. Through these connections, it is likely that the vestibular nuclei play a role in the conscious appreciation of body position [95, 110].
The vestibular nuclei also have extensive reciprocal connections with the reticular formation. “The vestibuloreticular connections are presumably involved in vomiting and cardiovascular reactions observed on vestibular irritation” [95].
In a classic sense, the vestibulocerebellum is primarily thought to play a role in equilibrium. This is because lesions to the flocculonodular lobe can result in general dysequilibrium and vertigo [110]. However, the vestibular nuclei have widespread connections; therefore, lesions within the vestibulocerebellum may also result in conditions such as nystagmus and a variety of autonomic concomitants [95]. It is quite possible that similar symptoms could develop when -adequate mechanoreceptive input does not reach the vestibular nuclei and flocculonodular lobe.
Cerebral cortex
As mentioned in the cerebellum section, each area in the primary somatosensory region receives afferents from specific receptors [107]. Group Ia muscle spindle afferents project to area 3a. Cutaneous afferents project to area 3b. Joint afferents project to area 2. Area 1 receives input from both cutaneous and deep tissue receptors. It is well-known that the dorsal columns and medial lemniscus carry this mechanoreceptive information. The importance of such mechanoreceptor afferent input to the cerebral cortex is described by many neuroanatomists.
Carpenter states that although impulses generated in-neurons in the primary motor area (M I), the premotor area, and in the supplementary motor area (M -II) are responsible for movement, motor control, changes in muscle tone and maintenance of posture, these motor activities are initiated by inputs that arise from the thalamus, other cortical areas and peripheral receptors [107].
According to Masdeu and Brazis, sensorimotor control is carried out by various thalamic nuclei, such as the ventrolateral nucleus, which predominantly coordinate the finer distal motor movements – [106]: “The ventrolateral nucleus integrates input -from the cerebellum, basal ganglia and mechanoreceptors from the musculoskeletal system; it projects to the pericentral cortex or primary motor cortex (area 4). ” Wyke maintained that Type I joint receptors, which are similar to Ruffini spray endings, excite the paracentral and parietal regions of the cerebral cortex and make a significant contribution to the perceptual experiences of postural sensation and kinesthesis [96]. Clearly, appropriate mechanoreceptor afferent input is required by the cerebral cortex to perform a host of conscious and subconscious motor functions.
Other authors indicate that afferent input plays a role that extends beyond that of motor control. Both Guyton [12] and Nolte [110] indicated that if afferent signals are eliminated, the cerebrum would be incapable of functioning in a conscious manner and would actually approach a permanent state of coma.
A more precise anatomical look at the cerebral cortex reveals that somatosensory input plays an additional role in human function, such that mechanoreceptor input is actually needed to help us function as humans. The human neocortex accounts for more than 90% of the total cortical area [110] and is divided into six separate layers [107]. Layer 1, the molecular layer, is the most external. Layer II is the external granular layer. Layer III is the external pyramidal layer. Layer IV is the internal granular layer. Layer V is the internal pyramidal layer. Layer VI, the deepest layer, is known as the multiform layer.
Generally speaking, layers II and IV receive afferent input and layers III and V receive mainly efferent data [112]. Layer II receives cortical afferents and, depending on the location in the neocortex, layer IV receives afferents from somatosensory receptors, the medial geniculate body for audition and the lateral geniculate body for vision (see discussion below on primary sensory areas). Layer III projects ipsilaterally and contralaterally to other areas of the cerebral cortex via association fibers and commissural fibers. Layer V gives rise to corticostriate fibers, corticopontine fibers, corticobulbar fibers and corticospinal fibers [107]. Layer VI is also thought to be largely -efferent and to mainly project fibers to the thalamus. Cortical interneurons allow the various layers in a specific area to communicate with one another [112].
The term ‘great sensory pathways’ has often been used to describe the somatosensory, optic-and acoustic systems [95]. Each has an individual primary sensory area in the cerebral cortex. Area 17 in the occipital lobe is for-vision. Area 41 in the temporal lobe is for audition, and areas 3,1 and 2 in the parietal lobe are for sornatosensory input. Once afferent input is received in a specific primary sensory area, the information is integrated and then communicated to other parts of the brain via commissural and association fibers. The great majority of commissural fibers travel in the corpus callosum, which is thought to contain about 180 million fibers. The projections between homotopic regions are thought to be very precise. For example, the commissural connections of the primary and secondary sensory areas seem to be extremely specific [95].
Association fibers are ipsilateral, and four main fiber pathways are typically described. One passes through the cingulate gyms and is known as the cingulum. The superior longitudinal fasciculus connects the frontal lobe to the occipital lobe. The inferior longitudinal fasciculus connects the occipital lobe to the temporal lobe. The uncinate fasciculus passes from the temporal lobe to the frontal lobe [95, 110]. It should be understood that none of the association pathways are discrete, point-to-point pathways. Fibers freely enter and leave along the course of the pathways [110]. Thus, the association pathways allow for an almost unimaginable number of connections. We know that the primary sensory areas (for somatosensory input, vision and audition) all communicate in various association areas, such as the parieto-occipital-temporal association area and the prefrontal association area, via the commissural and association pathways [95, 112].
The prefrontal association area also receives input from the limbic system; thu-s, this association area receives information about all sensory modalities as well as information regarding motivational and emotional states [112]. All of this input is integrated, which allows one to appropriately engage a given environmental situation. A reduction in limbic input, visual input, auditory input or somatosensory input would necessarily compromise–one’s ability to function.
Janse provided a most elegant commentary on the importance of mechanoreceptor afferent input [116]: Numerically, the somatic sensory factors comprise by far the major activating vehicle of the nervous system, and the overtones of the body’s conduct are significantly conditioned and controlled by their input.” Regarding the total sensorial experience of humans, which includes mechanoreceptive, nociceptive and special senses, Janse stated that humans are provided “with sentiencies [sic] of emotional, mental and spiritual affectivities that defy total comprehension.” Janse suggested that an divergence in the totality of sensory input could ultimately result in pathology, or symptoms of pathology, in seemingly unrelated tissues and organs [116].
Symptoms associated with reduced mechanoreceptor input
Mechanoreceptive information reaches numerous centers in the CNS. Consequently, a reduction in mechanoreceptor input caused by joint complex dysfunction has the potential to promote numerous symptoms that could mimic lesions of the vestibular nuclei, cerebellum, cerebral cortex and basal ganglia. Although much more research is still needed in this area, evidence exists to support this contention. For example, as early as 1845, “Longet reported that surgical damage of neck muscles in a wide range of species led to generalized but transient motor disturbances characterized by an ataxia similar to that which followed cerebellectomy” [103]. More recently, a study of patients with soft tissue injuries in the neck led the authors to conclude that oculomotor abnormalities may be caused by abnormal mechanoreceptor input [117].
Fitz-Ritson found that 112 of 235 patients with cervical spine tmuma experienced cervical vertigo [118]. The definition of vertigo used in this study included both “subjective vertigo, i.e., the patient feels that he/she is rotating, [and] objective vertigo, i.e., the feeling that the room or environment is rotating.” After 18 treatments, which involved chiropractic adjustments to restore mobility to restricted joints, 101 of the 112 patients (90.2%) were symptom-free. This finding is consistent with the findings of Lewit, who stated that manipulation is very effective for reducing vertigo and dizziness [119].
It seems that symptoms of enhanced nociception often accompany symptoms associated with reduced mechanoreception. For example, Weeks and Travell demonstrated that a clinical syndrome characterized by postural vertigo or dizziness, imbalance and, usually, headache may be caused by dysfunction of the sternocleidomastoid muscle [120]. Gray reported on a series of case histories that described how injured cervical muscles can play a role in the production of vertigo, pain, nausea and tinnitus [121].
In a article titled Cervical Vertigo, Wing and HargraveWilson described 80 patients, all of whom had some form of vertigo, varying from the severe rotary type to generalized unsteadiness [122]. All patients had a thorough examination of the ears, nose and throat, and the results were normal in 96% of the cases. Electronystagmographic abnormalities were found in all patients. Some 69% of the patients also had either occipital headaches or cervical pain. All patients had “tender muscle guarding” in the upper cervical region, which was thought to exist because of “partial fixation of the involved vertebra.” The authors stated that manipulation was a “bastion” of treatment. After treatment, electronystagmographic recordings were significantly improved in 73% of the patients. A total of 53% of patients had complete relief of symptoms, and 36% had significant improvement to the point where they required no medication and could return to normal activities.
Several additional authors have discussed the relationship between a dysfunctional cervical spine and symptoms of pain, vertigo and dizziness [123-126]. Research also suggests that joint complex dysfunction in the lumbar spine can affect equilibrium [127].
Reduced mechanoreception may also impact the nonmotor functions of the cerebellum. In 1978, Watson explained how traditional concepts of cerebellar physiology emphasize motor control functions; however, he also points out that an emerging body of literature demonstrates a relationship between the cerebellum and psychological processes [128].
Specifically, data have suggested that this brain strucmre may participate in sensory integration activities, motor skills learning, visual and auditory discrimination performance, emotion and motivation control, and reinforcement processes [128].
Many articles have discussed the cerebellum’s participation in emotional expression [128-132]. The most dramatic work was completed by Heath during the 1970s [132].
Through experimentation, Heath demonstrated that brain sites for emotional expression are anatomically connected and functionally related to sensory relay nuclei for all modalities and also to sites involved in facial expression and motor coordination [132]. His research demonstrated that efferent pathways from the vermis/fastigial nucleus could stimulate pleasure centers located in the septal nuclei of the hypothalamus and corticomedial amygdala, and simultaneously inhibit adversive emotion centers located in the hippocampus and dorsolateral amygdala. Heath applied electrical stimulation to the vermis to activate this pathway in his treatment of psychiatric patients. To access the cerebellum, suboccipital craniotomy was performed and 2-mm electrodes were implanted subtentorially over the rostral vermal and paravermal regions of the cerebellum. A pace-making device delivered a stimulus at selected time intervals. Heath used this method on 11 patients with intractable psychiatric illness, all of whom were pronounced incurable by at least two physicians. The length of illness varied from 6 to 23 yr. Of the 11 patients, four had uncontrollable violence-aggression (two with no demonstrable organic brain disease and two with brain pathology), five were schizophrenics, and two had lifetime patterns of severe neurosis. After treatment, 10 of the 11 patients were out of the hospital and functioning without medication or other treatment [except for cerebellar stimulationT. Some were symptom-free, and others demonstrated significant improvements. The one patient who failed to respond had an adhesion between the tentorium and the rostral vermis that extended 2.5 cm to either side of midline, which apparently damaged the targeted cerebellar neurons.
Heath’s work is important for chiropractors because the rostral vermal and paravermal neurons of the cerebellum receive axial and appendicular mechanoreceptor input from all levels of the spinal cord (see earlier discussion on the cerebellum). Although this does not prove that dysafferentation caused by joint complex dysfunction is a cause of psychiatric illness, it is clear that interesting implications do exist and should be investigated.
CONCLUSION
The information presented in this article demonstrates that the CNS is greatly influenced-by somatosensory input. Numerous, seemingly unrelated symptoms can be generated when nociceptive input is enhanced and mechanoreceptive input is reduced. Research evidence leads us to believe that such dysafferent input is associated with joint complex dysfunction, which explains why so many seemingly bizarre symptoms respond to chiropractic care.
As stated earlier, according to classic neuroanatomy, the great sensory systems include the visual system, the auditory system and the somatosensory system. At the present time, there are specialists devoted to -the visual and auditory systems. As of yet, no profession has effectively stepped forward to specialize in treating somatosensory system dysfunction (i.e., dysafferentation induced by joint complex dysfunction). That chiropractic has not assumed such a role is surprising. Indeed, it seems that Janse envisioned chiropractors filling this role [116].
Apparently, Janse was so convinced that the chiropractic profession would pursue research in the field of somatosensory neurology that he wrote, “Let us be a trifle bold and quite foolish; let us try to imagine what a symposium on Principles of Sensory Communication might be like in 10 or 15 years hence” [116]. Janse went on to discuss numerous aspects of somatosensory function that could have served as research topics for chiropractors. Unfortunately, save for a few scattered articles, the idea that chiropractors should be the doctors or care takers of one of the great sensory systems has not been pursued. Janse warned future generations of chiropractors about neglecting the somatosensory system. He stated:
It is not to be forgotten that essentiatly man is a sensorial organism. Re ftinctions by vintue of the in-puts he experiences via the cutaneum, the subdermal, the myofascial planes, the dianthrodial complexes of the musculoskeletal system, especially the spine; the related proprioceptive phenomena [116].
Suggestions for Chiropractic Education
At the present time, the basic sciences-in most chiropractic colleges are taught in the same fashion as in medical school. Students are taught to view anatomy, physiology and diagnosis based solely on a pathology model. In other words, a differential diagnosis involves ruling out pathological changes in anatomical structures and metabolic pathways. This approach should not be abandoned; however, joint complex dysfunction and dysafferentation must be included in every differential diagnosis whenever it is determined that nociceptive and mechanoreceptive input can directly or indirectly influence the structures from which the symptoms are generated. This demands that students and practitioners be well versed in the details of the central connections of the many neuroanatomical pathways related to nociceptive and mechanoreceptive input.
Without this knowledge, it will be impossible for students and doctors to understand and explain how joint complex dysfunction can affect the many conditions that afflict the human body.
At the present time, only a few articles explain how to include joint complex dysfunction in the differential diagnosis, all of which focus on the topic of dizziness and vertigo [99, 118, 133, 134]. This must change if chiropractic is to ever become a truly mainstream profession that reaches a majority of the population.
REFERENCES:
-
Seaman D. Joint complex dysfunction, a novel term to replace subluxation/subluxation complex: Etiological and treatment considerations J Manipulative Physiol Ther 1997; 20:634-44.
-
Waagen G, Strang V. Origin and development of traditional chiropractic philosophy. In: Haldeman S, editor. Principles and practice of chiropractic. 2nd ed. Norwalk: Appleton & Lange;1992. p.29-43.
-
Practice guidelines for straight chiropractic. Proceedings of the International Straight Chiropractic Consensus Conference. Chandler (AZ): World Chiropractic Alliance; 1992. p.98.
-
Kale Clinic & Research Center. Houston control (patient pamphlet). Spartanburg: South Carolina; 1993.
-
Seaman D. Proprioceptor: an obsolete, inaccurate word. J Manipulative Physiol Ther 1997; 20:279-84.
-
Seaman D. Letter to the editor. Top Clin Chiropr 1997; 4(1)
-
Cramer G, Darby S. Letter to the editor. Top Clin Chiropr 1997;4(1 ):viii-xi.
-
Deafferentation. Dorland’s Medical Dictionary. 25th ed. Philadelphia: WB Saunders; 1974. p.409.
-
Burchiel K. Deafferentation syndromes and dorsal root entry zone lesions. In: Fields H, editor. Pain syndromes in neurology. Boston: Butterworth-Heinemann; 1990. p.201-22.
-
Fields H. Pain. New York: McGraw-Hill; 1987.
-
Tasker R. Management of nociceptive, deafferentation and central pain by surgical intervention. In: Fields H, editor. Pain syndrome in neurology. Boston: Butterworth-Heinemann; 1990. p.143-200.
-
Guyton A. Basic neuroscience. 2nd ed. Philadelphia: WB Saunders; 1991.
-
Martin J, Jessell T. Modality coding in the somatic sensory system. In: Kandel E, Schwartz J, Jessell T, eds. Principles of neural science. 3rd ed. New York: Elsevier; 1991. p.341-52.
-
Willis W, Coggeshall R. Sensory mechanisms of the spinal cord. 2nd ed. New York: Plenum Press; 1991.
-
Schaible H, Schmidt R. Direct observation of the sensitization of articular afferents during an experimental arthritis. In: Dubner R, Gebhart G, Bond M, editors. Proceedings of the Fifth World Congress on Pain: 1987 Aug 2-7; Hamburg, Germany. New York: Elsevier; 1988. p.44-SO.
-
Guilbaud G. Peripheral and central electrophysiological mechanisms of joint muscle pain. In: Dubner R, Gebhart G, Bond M, editors. Proceedings of the Fifth World Congress on Pain; 1987 Aug 2-7; Hamburg, Germany. New York: Elsevier; 1988. p. 201-15.
-
Hanesch U, Heppelmann B, Messlinger K, Schmidt R. Noci-ception in normal and arthritic joints: structural and functional aspects. In: Willis W, editor. Hyperalgesia and allodynia. New York: Raven Press; 1992. p.81-106.
-
Schaible H, Grubb B. Afferent and spinal mechanisms of joint pain. Pain 1993; 55:5-54.
-
Schmidt R, Schaible H Messlinger K. Heppelmann B, Hanesch U, Pawlak M. Silent and active nociceptors: structure, functions, and clinical implications. In: Gebhart G, Hammond T, Jensen T, editors. Proceedings of the 7th World Congress on Pain:
-
Progress in Pain Research and Management; 1993; Paris, France. Seattle: IASP Press; 1994. p.213-SO.
-
Hooshmand H. Chronic pain: reflex sympathetic dystrophy, prevention and management. Boca Raton (FL): CRC Press; 1993. p.33-55.
-
Jozsa L, Balint J, Kannus P, Jarvinen M, Lehto M. Mechanoreceptors in human myotendinous junction. Muscle Nerve 1993; 16:453-7.
-
Lephart S. Reestablishing proprioception, kinesthesia, joint p0sition sense, and neuromuscular control in rehabilitation. In: Prentice W, ed. Rehabilitation techniques in sports medicine. 2nd ed. St. Louis: Mosby; 1994. p.118-37.
-
Parkhurst T, Burnett C. Injury and proprioception in the lower back. J Orthop Sports Phys Ther 1994; 19:282-95.
-
Slosberg M. Effects of altered afferent input on sensation, proprioception, muscle tone and sympathetic reflex response. J Manipulative Physiol Ther 1988; 11:400-8.
-
Jones M, Christensen N, Carr J. Clinical reasoning in orthopedic manual therapy. In: Grant R, editor. Physical therapy of cervical and thoracic spine. 2nd ed. New York: Churchill-Livingstone; 1994. p.89-108.
-
Peterson D. Principles of adjustive technique. In: Bergmann T, Peterson D, Lawrence D, editors. Chiropractic technique. New York: Churchill-Livingstone; 1993. p.123-95.
-
Peterson D, Bergrnann T. Joint assessment principles and procedures. In: Bergmann T, Peterson D, Lawrence D, editors. Chiropractic technique. New York: Churchill Livingstone; 1993. p.51-121.
-
Henderson C. Three neurophysiologic theories on the chiropractic subluxation. In: Gatterman M, editor. Foundations of chiropractic-subluxation. St. Louis: Mosby; 1995. p.225-33.
-
Murphy D. The locomotor system: Korr’s “primary machinery of life.” J Manipulative Physiol Ther 1994; 17:562-4.
-
Denslow J, Hassett C. The central excitatory state associated with postural abnormalities. J Neurophysiol 1942; 5:393-402.
-
Korr I. The neural basis of the osteopathic lesion. J Am Osteopath Assoc 1947; 47:191-7.
-
Patterson M. A model mechanism for spinal segmental facilitation. J Am Osteopath Assoc 1976; 76:62-72.
-
Miller B, Keane C, editors. Encyclopedia and dictionary of medicine, nursing and allied health. Philadelphia: WB Saunders; 1987. p.383-8.
-
Bogduk N. Innervation patterns of the cervical spine. In: Grant R, editor. Physical therapy of the cervical and thoracic spine. 2nd ed. New York: Churchill-Livingstone; 1994. p.65-76.
-
Bogduk N, Valencia F. Innervation patterns of the thoracic spine. In: Grant R, editor. Physical therapy of the cervical and thoracic spine. 2nd ed. New York: Churchill-Livingstone; 1994. p.77-87.
-
Bogduk N. Innervation, pain patterns, and mechanisms of pain production. In: Twomey L, Taylor J, editors. Physical therapy of the low back. 2nd ed. New York: Churchill-Livingstone; 1994. p.93-109.
-
Charman R. Pain and nociception: mechanisms and modulation in sensory context. In: Boyling J, Palastanga N, editors. Grieve’s modern manual therapy: the vertebral column. 2nd ed. New York: Churchill-Livingstone; 1994. p.253-70.
-
Grieve G. Common vertebral joint problems. 2nd ed. New York: Churchill-Livingstone; 1988. p.51-2.
-
Haldeman S. The neurophysiology of pain. In: Haldeman S, editor. Principles and practice of chiropractic. 2nd ed. Norwalk (CT): Appleton-Century-Crofts; 1992. p.165-84.
-
Wyke B. The neurological basis of thoracic pain. Rheumatol Phys Med 1970; 10:356-67.
-
Wyke B. Neurological aspects of pain therapy. In: Swerdlow M, editor. The therapy of pain. Philadelphia: JB Lippincott; 1980. p.1-30.
-
Price D. Psychological and neural mechanisms of pain. New York: Raven Press; 1988. p.76-99.
-
McMahon S, Koltzenburg M. Novel classes of nociceptors: beyond Sherrington. Trends Neurosci 1990; 13:199-201.
-
Bonica J. Anatomic and physiologic basis of nociception and pain. In: Bonica J, editor. Management of pain. 2nd ed. Philadelphia: Lea & Febiger; 1990. p.28-94.
-
Bonica J. Definitions and taxonomy of pain. In: Bonica J, editor. The management of pain. 2nd ed. Philadelphia: Lea & Febiger; 1990. p.18-27.
-
Maciewicz R. Organization of pain pathways. In: Ashbury A, McKhann G, McDonald W, editors. Diseases of the nervous system: clinical neurobiology. Philadelphia: WB Saunders; 1992. p.849-57
-
Casey K. Nociceptors and their sensitization: overview. In: Willis W, editor. Hyperalgesia and allodynia. New York: Raven Press; 1992. p.13-S.
-
Randwerker H, Peeh P. Nociceptors: chemosensitivity and sensitization by chemical agents. In: Willis W, editor. Hyperalgesia and allodynia. New York: Raven Press; 1992. p.107-iS.
-
Steen KH, Reeh PW, Anton F, Handwerker HO. Protons selectively induce lasting excitation and sensitization to mechanical stimulation of nociceptors in rat skin, in vitro. J Neurosci 1992; 12:86-95.
-
Campbell J, Meyer R, Davis K, Raja S. Sympathetically maintained pain: a unifying hypothesis. In: Willis W, editor. Hyperalgesia and allodynia. New York: Raven Press; 1992. p.141-9.
-
Levine J, Coderre T, Basbaum A. The peripheral nervous system and the inflammatory process. In: Dubner R, Gebhart 0, Bond M, editors. Proceedings of the Fifth World Congress on Pain; 1987 Aug 2-7; Hamburg, Germany. New York: Elsevier; 1987. p.33-43.
-
Levine J, Taiwo Y, Heller P. Hyperalgesic pain: inflammatory and neuropathic. In: Willis W, editor. Hyperalgesia and allodynia. New York: Raven Press; 1992. p.117-23.
-
Rang H, Bevan S, Dray A. Chemical activation of nociceptive peripheral neurones. Br Med Bull 1991; 47:534-48.
-
Reeh P. Chemical excitation and sensitization of nociceptors. In: Urban L, editor. Cellular mechanisms of sensory processing. Berlin: Springer-Verlag; 1994. p.119-31.
-
Dubner R. Spinal cord neuronal plasticity: mechanisms of persistent pain following tissue damage and nerve injury. In: Vecchiet D, Albe-Fessard D, Lindbolm U, Giamberardino M, editors. New trends in referred pain and hyperalgesia. New York: Elsevier; 1993. p.109-17.
-
Coderre T, Katz J, Vaccarino A, Melzack R. Contribution of central neuroplasticity to pathological pain: review of clinical and experimental evidence. Pain 1993; 52:259-85.
-
Light A. The initial processing of pain and its descending control: spinal and trigeminal systems. New York: Karger; 1992. p.28.
-
Woolf C. Physiological, inflammatory and neuropathic pain. Adv Tech Stand Neurosurg 1987; 15:39-62.
-
Denslow 1, Korr I, Krems A. Quantitative studies of chronic facilitation in human motoneurons. Am J Physiol 1947; 150: 229-38.
-
Patterson M. The spinal cord: participant in disorder. Spinal Manipulation 1993; 9(3):2-11.
-
Korr I. Symposium on the functional implications of segmental facilitation: a research report. J Am Osteopath Assoc 1955; 54:265-82.
-
Korr I. The facilitated segment: a factor in injury to the body framework. Osteopath Ann 1973; 1:10-12, 17-18.
-
Korr I. Andrew Taylor Still memorial lecture: research and practice-a century later. J Am Osteopath Assoc 1974; 73:362-70.
-
Korr I. Proprioceptors and somatic dysfunction. I Am Osteopath Assoc 1975; 74:638-50.
-
Korr I. Sustained sympathicotonia as a factor in disease. In: Korr I, editor. The neurobiologic mechanisms in manipulative therapy. New York: Plenum Press; 1978. p.229-68.
-
Korr I. Somatic dysfunction, osteopathic manipulative treatment, and the nervous system: a few facts, some theories, many questions. I Am Osteopath Assoc 1986; 86:109-14.
-
Korr I. Osteopathic research: the needed paradigm shift. I Am Osteopath Assoc 1991; 91:156-71.
-
Woolf C. Evidence for a central component of post-injury pain hypersensitivity. Nature 1983; 306:686-8.
-
Dubner R. Neuronal plasticity in the spinal and medullary dorsal horns: a possible role in central pain mechanisms. In: Casey K. Pain and central nervous system disease: the central pain syndromes. New York: Raven Press; 1991. p.143-55.
-
Dubner R, Ruda M. Activity-dependent neuronal plasticity following tissue injury and inflammation. Trends Neurosci 1992; 15:96-103.
-
Dubner R, Basbaum A. Spinal cord plasticity following tissue or nerve injury. In: Wall R, Melzack R, editors. Textbook of pain. 3rd ed. New York: Churchill-Livingstone; 1994. p.225-41.
-
Hoheisel U, Mense S. Long-term changes in discharge behavior of cat dorsal horn neurones following noxious stimulation of deep tissues. Pain 1989; 36:239-47.
-
Ren K, Rylden 3, Williams G, Ruda M, Dubner R. The effects of a non-competitive NMDA receptor antagonist, MK-801, on behavioral hyperalgesia and dorsal horn neuronal activity in rats with unilateral inflammation. Pain 1992; 50:331-44.
-
Richmond C, Bromley L, Woolf C. Preoperative morphine pre-empts postoperative pain. Lancet 1993; 342:73-5.
-
Roberts W. A hypothesis on the physiological basis for causalgia and related pains. Pain 1986; 24:297-311.
-
Wall P, Woolf C. Muscle but not cutaneous C-afferent input produces prolonged increases in the excitability of the flexion reflex in the rat. I Physiol 1984; 356:443-58.
-
Woolf C. Long term alterations in the excitability of the flexion reflex produced by peripheral tissue injury in the chronic decerebrate rat. Pain 1984; 18:325-43.
-
Woolf C. Generation of acute pain: central mechanisms. Br Med Bull 1991; 47:523-33.
-
Woolf C. Excitability changes in central neurons following peripheral damage: role of central sensitization in the pathogenesis of pain. In: Willis W, editor. Hyperalgesia and allodynia. New York: Raven Press; 1992. p.221-243.
-
Woolf C. The dorsal horn: state-dependent sensory processing and the generation of pain. In: Wall P, Melzack R, editors. The textbook of pain. 3rd ed. New York: Churchill-Livingstone; 1994. p.101-12.
-
Simone D. Neural mechanisms of hyperalgesia. Curr Opin Neurobiol 1992; 2:479-83.
-
Gillette R. Spinal cord mechanisms of referred pain and related neuroplasticity. In: Gatterman M, editor. Foundations of chiropractic: subluxation. St. Louis: Mosby; 1995. p.279-301.
-
Huttenlocher P. Neural plasticity. In: Ashbury A, McKhann G, McDonald W, editors. Diseases of the nervous system: clinical neurobiology. Philadelphia: WB Saunders; 1992. p.63-71.
-
Kandel E. Cellular mechanisms of learning and the biological basis of individually. In: Kandel E, Schwartz I, Jessell T, editors. Principles of neural science. 3rd ed. New York:
-
Elsevier; 1991. p.1009-31.
-
Teyler T, Grover L. Forms of long-term potentiation induced by NMDA and non-NMDA receptor activation. In: Baudry M, Thompson R, Davis I, editors. Synaptic plasticity: molecular, cellular aspects. Cambridge (MA): MIT Press; 1993. p.73-86.
-
Bonica I. Introduction: semantic, epidemiologic, and educational issues. In: Casey K, editor. Pain and central nervous system disease: the central pain syndromes. New York: Raven Press; 1991. p.13-29.
-
Bonica 3. Clinical importance of hyperalgesia. In: Willis W, editor. Hyperalgesia and Allodynia. New York: Raven Press; 1992. p.17-43.
-
Berne R, Matthew L. Physiology. 3rd ed. St. Louis: Mosby Year Book; 1993. p.949-79.
-
Dilman V, Dean W. The neuroendocrine theory of aging and degenerative disease. Pensacola: The Center for Bio-Gerontology; 1992. p.23-30.
-
Feinstein B, Langton I, Iameson R, Schiller F. Experiments on pain referred from deep somatic tissues. I Bone loint Surg Am 1954; 36:981-97.
-
Nansel D, Szlazak M. Somatic dysfunction and the phenomenon of visceral disease simulation: a probable explanation for the apparent effectiveness of somatic therapy in patients presumed to be suffering from true visceral disease. I Manipulative Physiol Ther 1995; 18:379-97
-
Mendel T, Wink C, Zimny M. Neural elements in human cervical intervertebral discs. Spine 1992; 17:132-5.
-
Willis W. Central plastic responses to pain. In: Gebhart G, Hammond D, Iensen T, editors. Proceedings of the 7th World Congress on Pain; 1993; Paris, France. Seattle: IASP Press; 1994. p.301-24.
-
De long P, de long I, Cohen B, Jongkees L. Ataxia and nystagmus induced by injection of local anesthetics in the neck. Ann Neurol 1977; 1:240-6.
-
Brodal A. Neurological anatomy. 3rd ed. New York: Oxford University Press; 1981.
-
Wyke B. Articular neurology and manipulative therapy. In: Glasgow E, Twomey L, Scull E, Kleynhans A, Idczak R, editors. Aspects of manipulative therapy. 2nd ed. New York: Churchill-Livingstone; 1985. p.72-7.
-
Gillette R. Potential antinociceptive effects of high level somatic stimulation- chiropractic manipulation therapy may coactivate both tonic and phasic analgesic systems. Some recent evidence. Trans Pac Consortium Res 1986; 1:A4(1)-A4(9).
-
Woolf C. Segmental afferent fibre-mediated analgesia: transcutaneous electrical nerve stimulation (TENS) and vibration. In:
-
Wall P, Melzack R, editors. Textbook of pain. 2nd ed. New York: Churchill-Livingstone; 1989. p.884-96.
-
Dvorak I, Dvorak V. Manual medicine: diagnostics. 2nd ed. New York: Thieme Medical Publishers; 1990.
-
Gordon I. Spinal mechanisms of motor coordination. In: Kandel E, Schwartz I, lessell T. editors. Principles of neural science. 3rd ed. New York: Elsevier; 1991. p.581-95.
-
Sato A. Spinal reflex physiology. In: Haldeman S, editor. Principles and practice of chiropractic. 2nd ed. Norwalk (CT): Appleton & Lange; 1992. p.87-103.
-
Fitz-Ritson D. The anatomy and physiology of the muscle spindle, and its role in posture and movement: review. ICCA 1982; 26:144-50.
-
Richmond F, Bakker D, Stacey M. The sensorium: receptors of neck muscles and joints. In: Peterson B, Richmond F, editors. Control of head movement. New York: Oxford University Press; 1988. p.49-62.
-
Bogduk N, Twomey L. Clinical anatomy of the lumbar spine. 2nd ed. New York: Churchill-Livingstone; 1991. p.86.
-
Proske U. The golgi tendon organ. In: Dyck P, Thomas P, Griffin I, Low P, Podusko I, editors. Peripheral neuropathy. 3rd ed. Philadelphia: WB Saunders; 1993. p.141-8.
-
Masdeu I, Brazis P. The anatomic localization of lesions in the thalamus. In: Brazis P, Masdeu I, Biller I, editors. Localization in clinical neurology. 2nd ed. Boston: Little, Brown and Company; 1990. p.319-43.
-
Carpenter M. Core text of neuroanatomy. 4th ed. Baltimore: Williams & Wilkins; 1991. p.224-49.
-
Lantz C. The vertebral subluxation complex. ICA Rev Chiropr 1989:37-61.
-
Lantz C. The vertebral subluxation complex. In: Gatterman M, editor. Foundations of chiropractic: subluxation. St. Louis: Mosby; 1995. p. 149-74.
-
Nolte 3. The human brain. 3rd ed. St. Louis: Mosby; 1993. p. 337-59.
-
Ghez C. The cerebellum. In: Kandel E, Schwartz 3, Jessell T, editors. Principles of neural science. 3rd ed. New York: Elsevier; 1991. p. 626-46.
-
Brodal P. The central nervous system: structure and function. New York: Oxford University Press; 1992.
-
Fitz-Ritson D. Neuroanatomy and neurophysiology of the upper cervical spine. In: Vernon H, editor. Upper cervical spine: chiropractic diagnosis and treatment. Baltimore: Williams & Wilkins; 1988. p.48-85.
-
Bakker D, Abrahams V. Central projections from nuchal afferent systems. In: Peterson B, Richmond F, editors. Control of head movement. New York: Oxford University Press; 1988. p. 63-89.
-
Maeda M. Neck influences on the vestibulo-ocular reflex arc and the vestibulocerebellum. Prog Brain Res 1979; 50:551-9.
-
Janse 3. The integrative purpose and function of the nervous system: a review of classical literature. 3 Manipulative Physiol Ther 1978; 1:182-91.
-
Hildingsson C, Wenngren B, Toolanen G. Eye motility after soft-tissue injury of the cervical spine: a controlled, prospective study of 38 patients. Acta Orthop Scand 1993; 64:129-32.
-
Fitz-Ritson D. Cervicogenic vertigo. 3 Manipulative Physiol Ther 1991; 14:193-8.
-
Lewit K. Manipulative therapy in rehabilitation of the locomotor system. 2nd ed. Boston: Butterworth Heinemann; 1991. p.18.
-
Weeks V, Travell 3. Postural vertigo due to trigger areas in the sternocleidomastoid muscle. 3 Pediatr 1957; 47:315-27.
-
Gray L. Extra labyrinthine vertigo due to cervical muscle lesions. 3 Laryngol Otol 1956; 70:352-61.
-
Wing L, Hargrave-Wilson W. Cervical vertigo. Aust N Z 3 Surg 1974; 44(3):275-7.
-
Caranasos G, Israel R. Gait disorders in the elderly. Hosp Pract 1991:67-94.
-
Revel M, Andre-Deshays C, Minguet M. Cervicocephalic kinesthetic sensibility in patients with cervical pain. Arch Phys Med Rehabil 1991; 72:288-91.
-
Revel M, Minguet M, Gergory P, Vaillant 3, Manuel 3. Changes in cervicocephalic kinesthesia after a proprioceptive rehabilitation program of in patients with neck pain: a randomized controlled study. Arch Phys Med Rehabil 1994; 75:895-9.
-
Hulse M. Disequilibrium, caused by a functional disturbance in the upper cervical spine: clinical aspects and differential diagnosis. Man Med 1983; 1:18-22.
-
Hinoki M, Ushio N. Lumbomuscular proprioceptive reflexes in body equilibrium. Acta Otolaryngol Suppl (Stockh) 1975; 330: 197-210.
-
Watson P. Nonmotor functions of the cerebellum. Psychol Rev 1978; 85:944-67.
-
Courchesne E, Yeung-Courchesne R, Press G, Hesselink J, Jernigan T. Hypoplasia of cerebellar vermal lobules VI and VII in autism. N Engl 3 Med 1988; 318:1349-54.
-
Gutzmann H, Kuhl K. Emotion control and cerebellar atrophy in senile dementia. Arch Gerontal Geriatr 1987; 6:61-71.
-
Lalonde R, Botez M. The cerebellum and learning processes in animals. Brain Res Rev 1990; 15:325-32.
-
Heath R. Modulation of emotion with a brain pacemaker. 3 Nerv Ment Dis 1977; 165:30047.
-
Douglas F. The dizzy patient: strategic approach to history, examination, diagnosis, and treatment. Chiropr Technique 1993; 5:5-14.
-
Schimp D. A diagnostic algorithm for the dizzy patient. Chiropr Technique 1994; 6:123-37.




